Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from January, 2014
Show All
Posted by
Varun C N
January 28, 2014
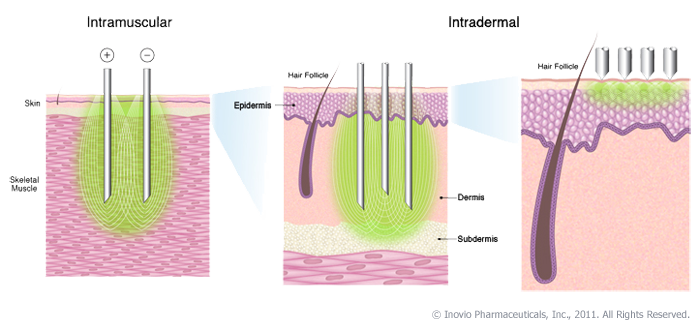
NextGen vaccination: Needle free devices, Interleukin adjuvants
Posted by
Varun C N
January 25, 2014
Clostridium difficile on a note
Posted by
Varun C N
January 15, 2014
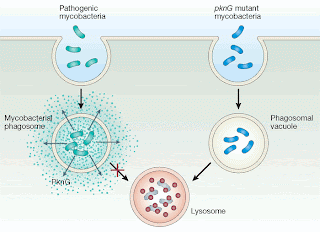
PDIM: Yet another virulence factor of TB
Posted by
Varun C N
January 06, 2014
Bacterial Persistence- On a Note
Newer Posts
Older Posts
Home