Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from June, 2016
Show All
Posted by
Varun C N
June 30, 2016
BtB#7: Immune cells of Central Nervous System
Posted by
Varun C N
June 27, 2016
HIV nuclear entry
Posted by
Varun C N
June 23, 2016
NGS for Microbial Diagnostics
Posted by
Varun C N
June 15, 2016
VDPV in Sewer sample- Implications
Posted by
Varun C N
June 13, 2016
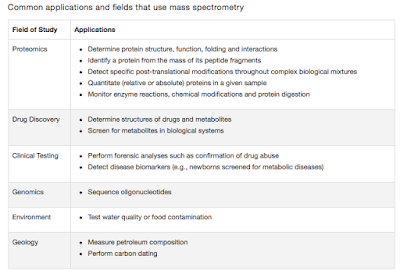
Lab series# 13: Mass Spectrometry Instrumentation Principles
Posted by
Varun C N
June 10, 2016
Lab Series# 12: Cryo-Preservation of cells
Posted by
Varun C N
June 06, 2016
Totally Drug Resistant Tuberculosis is actually XDR Plus
Posted by
Varun C N
June 03, 2016
First case of MCR-1 in US
Newer Posts
Older Posts
Home