Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from March, 2015
Show All
Posted by
Varun C N
March 28, 2015
Quasi Species: Defending my follow up
Posted by
Varun C N
March 26, 2015
Cerebral malaria- In short
Posted by
Varun C N
March 23, 2015
Quasi Species- Discussion
Posted by
Varun C N
March 13, 2015
Indian Influenza Outbreak: 2015- Follow up
Posted by
Varun C N
March 09, 2015
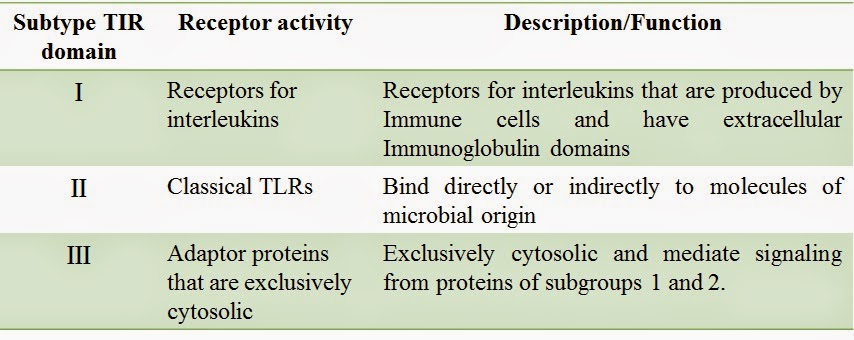
Toll Like Receptors
Posted by
Varun C N
March 03, 2015
A thought on Microbiome
Newer Posts
Older Posts
Home