Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from April, 2015
Show All
Posted by
Varun C N
April 30, 2015
Blogger's Desk #6: Spark of Ethical Debate on editing the embryonic genome
Posted by
Varun C N
April 29, 2015
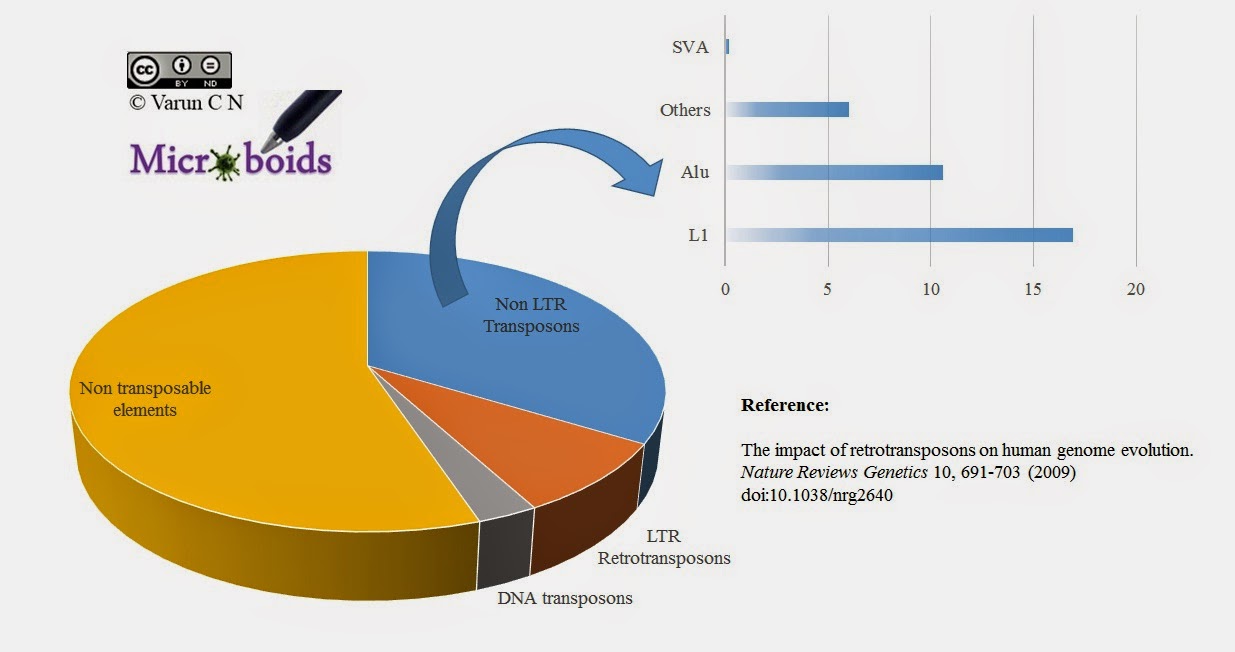
Retroviral protection
Posted by
Varun C N
April 28, 2015
Lab Series #3: Flow Cytometry
Posted by
Varun C N
April 20, 2015
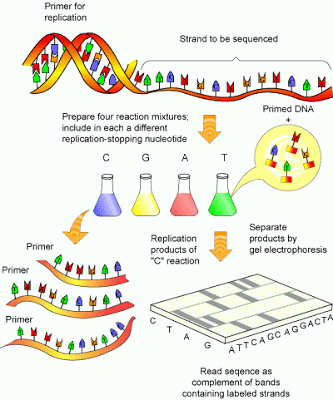
Lab Series #2: DNA sequencing
Posted by
Varun C N
April 19, 2015
Lab Series #1: Luminescence
Newer Posts
Older Posts
Home