Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from September, 2015
Show All
Posted by
Varun C N
September 30, 2015
Lab Series# 7: O&P examination of Stool Samples
Posted by
Varun C N
September 26, 2015
Wanted suspect: L pneumophilia
Posted by
Varun C N
September 23, 2015
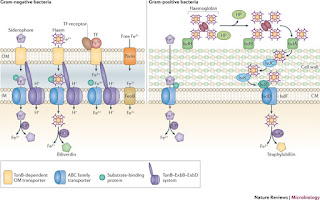
Will go to war for Iron: Bacterial strategy
Posted by
Varun C N
September 15, 2015
BtB#4- Sterilization/Disinfection in brief
Posted by
Varun C N
September 07, 2015
Ebola 2014: Not yet over in 2015!!
Posted by
Varun C N
September 01, 2015
Blogger's Desk #8- Statistics in Layman's language
Newer Posts
Older Posts
Home