Skip to main content
Search
Search
Microboids
Sharing Knowledge improves Knowledge... Knowledge should come at as less cost as possible.
Blog
Posts
More…
Posts
Showing posts from September, 2013
Show All
Posted by
Varun C N
September 30, 2013
HIV restriction factor- Mx2
Posted by
Varun C N
September 29, 2013
HIV Toolbox
Posted by
Varun C N
September 20, 2013
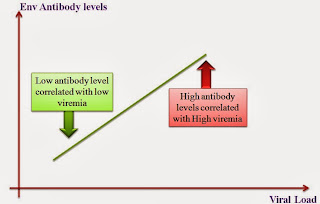
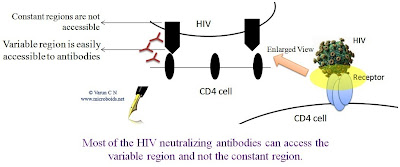
Critical analysis of HIV vaccines- Part II
Posted by
Varun C N
September 19, 2013
Critical analysis of HIV vaccines- Part I
Posted by
Varun C N
September 12, 2013
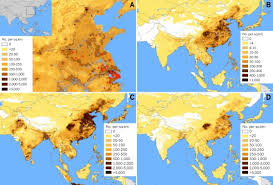
Headlines- MERS and MERS vaccine
Posted by
Varun C N
September 04, 2013
SAM vaccine for H7N9
Posted by
Varun C N
September 02, 2013
No Tit for HIV TAT
Newer Posts
Older Posts
Home